Fludarabine: Package Insert / Prescribing Info
Package insert / product label
Generic name: fludarabine phosphate
Dosage form: injection, solution
Drug class: Antimetabolites
J Code (medical billing code): J9185 (50 mg, injection)
Medically reviewed by Drugs.com. Last updated on Oct 25, 2024.
On This Page
WARNING
Fludarabine should be administered under the supervision of a qualified physician experienced in the use of antineoplastic therapy. Fludarabine can severely suppress bone marrow function. When used at high doses in dose-ranging studies in patients with acute leukemia, fludarabine was associated with severe neurologic effects, including blindness, coma, and death. This severe central nervous system toxicity occurred in 36% of patients treated with doses approximately four times greater (96 mg/m 2/day for 5 to 7 days) than the recommended dose. Similar severe central nervous system toxicity has been rarely (0.2%) reported in patients treated at doses in the range of the dose recommended for chronic lymphocytic leukemia.
Instances of life-threatening and sometimes fatal autoimmune hemolytic anemia have been reported to occur after one or more cycles of treatment with fludarabine. Patients undergoing treatment with fludarabine should be evaluated and closely monitored for hemolysis.
In a clinical investigation using fludarabine in combination with pentostatin (deoxycoformycin) for the treatment of refractory chronic lymphocytic leukemia (CLL), there was an unacceptably high incidence of fatal pulmonary toxicity. Therefore, the use of fludarabine in combination with pentostatin is not recommended.
Fludarabine Description
Fludarabine Phosphate Injection, USP contains fludarabine phosphate, a fluorinated nucleotide analog of the antiviral agent vidarabine, 9-ß-D-arabinofuranosyladenine (ara-A) that is relatively resistant to deamination by adenosine deaminase. Each mL contains 25 mg of the active ingredient fludarabine phosphate, 25 mg of mannitol, water for injection, q.s., and sodium hydroxide to adjust pH to 6.8. The pH range for the final product is 6.0 to 7.1. Fludarabine Phosphate Injection, USP is a sterile solution intended for intravenous administration.
The chemical name for fludarabine phosphate is 9 H-Purin-6-amine, 2-fluoro-9-(5- O-phosphono-ß-D-arabinofuranosyl) (2-fluoro-ara-AMP). The structure is:
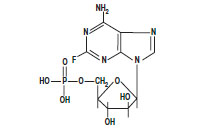
C 10H 13FN 5O 7P M.W. 365.2
Fludarabine - Clinical Pharmacology
Fludarabine phosphate is rapidly dephosphorylated to 2-fluoro-ara-A and then phosphorylated intracellularly by deoxycytidine kinase to the active triphosphate, 2-fluoro-ara-ATP. This metabolite appears to act by inhibiting DNA polymerase alpha, ribonucleotide reductase and DNA primase, thus inhibiting DNA synthesis. The mechanism of action of this antimetabolite is not completely characterized and may be multi-faceted.
Phase I studies in humans have demonstrated that fludarabine phosphate is rapidly converted to the active metabolite, 2-fluoro-ara-A, within minutes after intravenous infusion. Consequently, clinical pharmacology studies have focused on 2-fluoro-ara-A pharmacokinetics. After the five daily doses of 25 mg 2-fluoro-ara-AMP/m 2 to cancer patients infused over 30 minutes, 2-fluoro-ara-A concentrations show a moderate accumulation. During a 5-day treatment schedule, 2-fluoro-ara-A plasma trough levels increased by a factor of about 2. The terminal half-life of 2-fluoro-ara-A was estimated as approximately 20 hours. In vitro, plasma protein binding of fludarabine ranged between 19% and 29%.
A correlation was noted between the degree of absolute granulocyte count nadir and increased area under the concentration x time curve (AUC).
Special Populations
Pediatric Patients
Limited pharmacokinetic data for fludarabine phosphate for injection are available from a published study of children (ages 1 to 21 years) with refractory acute leukemias or solid tumors (Children’s Cancer Group Study 097). When fludarabine phosphate for injection was administered as a loading dose over 10 minutes immediately followed by a 5-day continuous infusion, steady-state conditions were reached early.
Patients with Renal Impairment
The total body clearance of the principal metabolite 2-fluoro-ara-A correlated with the creatinine clearance, indicating the importance of the renal excretion pathway for the elimination of the drug. Renal clearance represents approximately 40% of the total body clearance. Patients with moderate renal impairment (17 to 41 mL/min/m 2) receiving 20% reduced fludarabine dose had a similar exposure (AUC; 21 versus 20 nM•h/mL) compared to patients with normal renal function receiving the recommended dose. The mean total body clearance was 172 mL/min for normal and 124 mL/min for patients with moderately impaired renal function.
Clinical Studies
Two single-arm open-label studies of fludarabine have been conducted in adult patients with CLL refractory to at least one prior standard alkylating-agent containing regimen. In a study conducted by M.D. Anderson Cancer Center (MDAH), 48 patients were treated with a dose of 22 to 40 mg/m 2 daily for 5 days every 28 days. Another study conducted by the Southwest Oncology Group (SWOG) involved 31 patients treated with a dose of 15 to 25 mg/m 2 daily for 5 days every 28 days. The overall objective response rates were 48% and 32% in the MDAH and SWOG studies, respectively. The complete response rate in both studies was 13%; the partial response rate was 35% in the MDAH study and 19% in the SWOG study. These response rates were obtained using standardized response criteria developed by the National Cancer Institute CLL Working Group and were achieved in heavily pre-treated patients. The ability of fludarabine to induce a significant rate of response in refractory patients suggests minimal cross-resistance with commonly used anti-CLL agents.
The median time to response in the MDAH and SWOG studies was 7 weeks (range of 1 to 68 weeks) and 21 weeks (range of 1 to 53 weeks) respectively. The median duration of disease control was 91 weeks (MDAH) and 65 weeks (SWOG). The median survival of all refractory CLL patients treated with fludarabine was 43 weeks and 52 weeks in the MDAH and SWOG studies, respectively.
Rai stage improved to Stage II or better in 7 of 12 MDAH responders (58%) and in 5 of 7 SWOG responders (71%) who were Stage III or IV at baseline. In the combined studies, mean hemoglobin concentration improved from 9 g/dL at baseline to 11.8 g/dL at the time of response in a subgroup of anemic patients. Similarly, average platelet count improved from 63,500/mm 3 to 103,300/mm 3 at the time of response in a subgroup of patients who were thrombocytopenic at baseline.
Indications and Usage for Fludarabine
Fludarabine Phosphate Injection, USP is indicated for the treatment of patients with B-cell chronic lymphocytic leukemia (CLL) who have not responded to or whose disease has progressed during treatment with at least one standard alkylating-agent containing regimen. The safety and effectiveness of Fludarabine Phosphate Injection, USP in previously untreated or non-refractory patients with CLL have not been established.
Contraindications
Fludarabine Phosphate Injection, USP is contraindicated in those patients who are hypersensitive to this drug or its components.
Warnings
There are clear dose-dependent toxic effects seen with fludarabine. Dose levels approximately 4 times greater (96 mg/m 2/day for 5 to 7 days) than that recommended for CLL (25 mg/m 2/day for 5 days) were associated with a syndrome characterized by delayed blindness, coma and death. Symptoms appeared from 21 to 60 days following the last dose. Thirteen of 36 patients (36%) who received fludarabine at high doses (96 mg/m 2/day for 5 to 7 days) developed this severe neurotoxicity. This syndrome has been reported rarely in patients treated with doses in the range of the recommended CLL dose of 25 mg/m 2/day for 5 days every 28 days. The effect of chronic administration of fludarabine on the central nervous system is unknown; however, patients have received the recommended dose for up to 15 courses of therapy.
Severe bone marrow suppression, notably anemia, thrombocytopenia and neutropenia, has been reported in patients treated with fludarabine. In a Phase I study in solid tumor patients, the median time to nadir counts was 13 days (range, 3 to 25 days) for granulocytes and 16 days (range, 2 to 32) for platelets. Most patients had hematologic impairment at baseline either as a result of disease or as a result of prior myelosuppressive therapy. Cumulative myelosuppression may be seen. While chemotherapy induced myelosuppression is often reversible, administration of fludarabine requires careful hematologic monitoring.
Several instances of trilineage bone marrow hypoplasia or aplasia resulting in pancytopenia, sometimes resulting in death, have been reported. The duration of clinically significant cytopenia in the reported cases has ranged from approximately 2 months to approximately 1 year. These episodes have occurred both in previously treated or untreated patients.
Instances of life-threatening and sometimes fatal autoimmune hemolytic anemia have been reported to occur after one or more cycles of treatment with fludarabine in patients with or without a previous history of autoimmune hemolytic anemia or a positive Coombs’ test and who may or may not be in remission from their disease. Steroids may or may not be effective in controlling these hemolytic episodes. The majority of patients rechallenged with fludarabine developed a recurrence in the hemolytic process. The mechanism(s) which predispose patients to the development of this complication has not been identified. Patients undergoing treatment with fludarabine should be evaluated and closely monitored for hemolysis.
Transfusion-associated graft-versus-host disease has been observed rarely after transfusion of non-irradiated blood in fludarabine treated patients. Consideration should, therefore, be given to the use of irradiated blood products in those patients requiring transfusions while undergoing treatment with fludarabine.
In a clinical investigation using fludarabine in combination with pentostatin (deoxycoformycin) for the treatment of refractory chronic lymphocytic leukemia (CLL) in adults, there was an unacceptably high incidence of fatal pulmonary toxicity. Therefore, the use of fludarabine in combination with pentostatin is not recommended.
Of the 133 CLL adult patients in the two trials, there were 29 fatalities during study. Approximately 50% of the fatalities were due to infection and 25% due to progressive disease.
Pregnancy Category D
Fludarabine may cause fetal harm when administered to a pregnant woman. Fludarabine phosphate was teratogenic in rats and in rabbits. Fludarabine phosphate was administered intravenously at doses of 0, 1, 10 or 30 mg/kg/day to pregnant rats on days 6 to 15 of gestation. At 10 and 30 mg/kg/day in rats, there was an increased incidence of various skeletal malformations. Fludarabine phosphate was administered intravenously at doses of 0, 1, 5 or 8 mg/kg/day to pregnant rabbits on days 6 to 15 of gestation. Dose-related teratogenic effects manifested by external deformities and skeletal malformations were observed in the rabbits at 5 and 8 mg/kg/day. Drug-related deaths or toxic effects on maternal and fetal weights were not observed. There are no adequate and well-controlled studies in pregnant women.
If fludarabine is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
Precautions
General
Fludarabine is a potent antineoplastic agent with potentially significant toxic side effects. Patients undergoing therapy should be closely observed for signs of hematologic and nonhematologic toxicity. Periodic assessment of peripheral blood counts is recommended to detect the development of anemia, neutropenia and thrombocytopenia.
Tumor lysis syndrome associated with fludarabine treatment has been reported in CLL patients with large tumor burdens. Since fludarabine can induce a response as early as the first week of treatment, precautions should be taken in those patients at risk of developing this complication.
There are inadequate data on dosing of patients with renal insufficiency. Fludarabine must be administered cautiously in patients with renal insufficiency. The total body clearance of 2-fluoro-ara-A has been shown to be directly correlated with creatinine clearance. Patients with moderate impairment of renal function (creatinine clearance 30 to 70 mL/min/1.73 m 2) should have their fludarabine dose reduced by 20% and be monitored closely. Fludarabine is not recommended for patients with severely impaired renal function (creatinine clearance less than 30 mL/min/1.73 m 2).
Laboratory Tests
During treatment, the patient’s hematologic profile (particularly neutrophils and platelets) should be monitored regularly to determine the degree of hematopoietic suppression.
Drug Interactions
The use of fludarabine in combination with pentostatin is not recommended due to the risk of severe pulmonary toxicity (see WARNINGS).
Mutagenesis
Fludarabine phosphate was not mutagenic to bacteria (Ames test) or mammalian cells (HGRPT assay in Chinese hamster ovary cells) either in the presence or absence of metabolic activation. Fludarabine phosphate was clastogenic in vitro to Chinese hamster ovary cells (chromosome aberrations in the presence of metabolic activation) and induced sister chromatid exchanges both with and without metabolic activation. In addition, fludarabine phosphate was clastogenic in vivo (mouse micronucleus assay) but was not mutagenic to germ cells (dominant lethal test in male mice).
Impairment of Fertility
Studies in mice, rats and dogs have demonstrated dose-related adverse effects on the male reproductive system. Observations consisted of a decrease in mean testicular weights in mice and rats with a trend toward decreased testicular weights in dogs and degeneration and necrosis of spermatogenic epithelium of the testes in mice, rats and dogs. The possible adverse effects on fertility in humans have not been adequately evaluated.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from fludarabine, a decision should be made to discontinue nursing or discontinue the drug, taking into account the importance of the drug for the mother.
Pediatric Use
Data submitted to the FDA was insufficient to establish efficacy in any childhood malignancy. Fludarabine was evaluated in 62 pediatric patients (median age 10, range 1 to 21) with refractory acute leukemia (45 patients) or solid tumors (17 patients). The fludarabine regimen tested for pediatric acute lymphocytic leukemia (ALL) patients was a loading bolus of 10.5 mg/m 2/day followed by a continuous infusion of 30.5 mg/m 2/day for 5 days. In 12 pediatric patients with solid tumors, dose-limiting myelosuppression was observed with a loading dose of 8 mg/m 2/day followed by a continuous infusion of 23.5 mg/m 2/day for 5 days. The maximum tolerated dose was a loading dose of 7 mg/m 2/day followed by a continuous infusion of 20 mg/m 2/day for 5 days. Treatment toxicity included bone marrow suppression. Platelet counts appeared to be more sensitive to the effects of fludarabine than hemoglobin and white blood cell counts. Other adverse events included fever, chills, asthenia, rash, nausea, vomiting, diarrhea, and infection. There were no reported occurrences of peripheral neuropathy or pulmonary hypersensitivity reaction.
Adverse Reactions/Side Effects
The most common adverse events include myelosuppression (neutropenia, thrombocytopenia and anemia), fever and chills, infection, and nausea and vomiting. Other commonly reported events include malaise, fatigue, anorexia, and weakness. Serious opportunistic infections have occurred in CLL patients treated with fludarabine. The most frequently reported adverse events and those reactions which are more clearly related to the drug are arranged below according to body system.
Hematopoietic Systems
Hematologic events (neutropenia, thrombocytopenia, and/or anemia) were reported in the majority of CLL patients treated with fludarabine. During fludarabine treatment of 133 patients with CLL, the absolute neutrophil count decreased to less than 500/mm 3 in 59% of patients, hemoglobin decreased from pretreatment values by at least 2 grams percent in 60%, and platelet count decreased from pretreatment values by at least 50% in 55%. Myelosuppression may be severe, cumulative, and may affect multiple cell lines. Bone marrow fibrosis occurred in one CLL patient treated with fludarabine.
Several instances of trilineage bone marrow hypoplasia or aplasia resulting in pancytopenia, sometimes resulting in death, have been reported in postmarketing surveillance. The duration of clinically significant cytopenia in the reported cases has ranged from approximately 2 months to approximately 1 year. These episodes have occurred both in previously treated or untreated patients.
Life-threatening and sometimes fatal autoimmune hemolytic anemia have been reported to occur in patients receiving fludarabine (see WARNINGS). The majority of patients rechallenged with fludarabine developed a recurrence in the hemolytic process.
Metabolic
Tumor lysis syndrome has been reported in CLL patients treated with fludarabine. This complication may include hyperuricemia, hyperphosphatemia, hypocalcemia, metabolic acidosis, hyperkalemia, hematuria, urate crystalluria, and renal failure. The onset of this syndrome may be heralded by flank pain and hematuria.
Nervous System
(See WARNINGS).
Objective weakness, agitation, confusion, visual disturbances, and coma have occurred in CLL patients treated with fludarabine at the recommended dose. Peripheral neuropathy has been observed in patients treated with fludarabine and one case of wrist-drop was reported.
Pulmonary System
Pneumonia, a frequent manifestation of infection in CLL patients, occurred in 16% and 22% of those treated with fludarabine in the MDAH and SWOG studies, respectively. Pulmonary hypersensitivity reactions to fludarabine characterized by dyspnea, cough and interstitial pulmonary infiltrate have been observed.
In postmarketing experience, cases of severe pulmonary toxicity have been observed with fludarabine use which resulted in ARDS, respiratory distress, pulmonary hemorrhage, pulmonary fibrosis, and respiratory failure. After an infectious origin has been excluded, some patients experienced symptom improvement with corticosteroids.
Gastrointestinal System
Gastrointestinal disturbances such as nausea and vomiting, anorexia, diarrhea, stomatitis, and gastrointestinal bleeding have been reported in patients treated with fludarabine.
Cardiovascular
Edema has been frequently reported. One patient developed a pericardial effusion possibly related to treatment with fludarabine. No other severe cardiovascular events were considered to be drug related.
Genitourinary System
Rare cases of hemorrhagic cystitis have been reported in patients treated with fludarabine.
Skin
Skin toxicity, consisting primarily of skin rashes, has been reported in patients treated with fludarabine.
Data in the following table are derived from the 133 patients with CLL who received fludarabine in the MDAH and SWOG studies.
PERCENT OF CLL PATIENTS REPORTING
NON-HEMATOLOGIC ADVERSE EVENTS
| ADVERSE EVENTS
| MDAH
(N=101) | SWOG
(N=32) |
| ANY ADVERSE EVENT
| 88%
| 91%
|
| BODY AS A WHOLE
| 72
| 84
|
| FEVER
| 60
| 69
|
| CHILLS
| 11
| 19
|
| FATIGUE
| 10
| 38
|
| INFECTION
| 33
| 44
|
| PAIN
| 20
| 22
|
| MALAISE
| 8
| 6
|
| DIAPHORESIS
| 1
| 13
|
| ALOPECIA
| 0
| 3
|
| ANAPHYLAXIS
| 1
| 0
|
| HEMORRHAGE
| 1
| 0
|
| HYPERGLYCEMIA
| 1
| 6
|
| DEHYDRATION
| 1
| 0
|
| NEUROLOGICAL
| 21
| 69
|
| WEAKNESS
| 9
| 65
|
| PARESTHESIA
| 4
| 12
|
| HEADACHE
| 3
| 0
|
| VISUAL DISTURBANCE
| 3
| 15
|
| HEARING LOSS
| 2
| 6
|
| SLEEP DISORDER
| 1
| 3
|
| DEPRESSION
| 1
| 0
|
| CEREBELLAR SYNDROME
| 1
| 0
|
| IMPAIRED MENTATION
| 1
| 0
|
| PULMONARY
| 35
| 69
|
| COUGH
| 10
| 44
|
| PNEUMONIA
| 16
| 22
|
| DYSPNEA
| 9
| 22
|
| SINUSITIS
| 5
| 0
|
| PHARYNGITIS
| 0
| 9
|
| UPPER RESPIRATORY INFECTION
| 2
| 16
|
| ALLERGIC PNEUMONITIS
| 0
| 6
|
| EPISTAXIS
| 1
| 0
|
| HEMOPTYSIS
| 1
| 6
|
| BRONCHITIS
| 1
| 0
|
| HYPOXIA
| 1
| 0
|
| GASTROINTESTINAL
| 46
| 63
|
| NAUSEA/VOMITING
| 36
| 31
|
| DIARRHEA
| 15
| 13
|
| ANOREXIA
| 7
| 34
|
| STOMATITIS
| 9
| 0
|
| GI BLEEDING
| 3
| 13
|
| ESOPHAGITIS
| 3
| 0
|
| MUCOSITIS
| 2
| 0
|
| LIVER FAILURE
| 1
| 0
|
| ABNORMAL LIVER FUNCTION TEST
| 1
| 3
|
| CHOLELITHIASIS
| 0
| 3
|
| CONSTIPATION
| 1
| 3
|
| DYSPHAGIA
| 1
| 0
|
| CUTANEOUS
| 17
| 18
|
| RASH
| 15
| 15
|
| PRURITUS
| 1
| 3
|
| SEBORRHEA
| 1
| 0
|
| GENITOURINARY
| 12
| 22
|
| DYSURIA
| 4
| 3
|
| URINARY INFECTION
| 2
| 15
|
| HEMATURIA
| 2
| 3
|
| RENAL FAILURE
| 1
| 0
|
| ABNORMAL RENAL FUNCTION TEST
| 1
| 0
|
| PROTEINURIA
| 1
| 0
|
| HESITANCY
| 0
| 3
|
| CARDIOVASCULAR
| 12
| 38
|
| EDEMA
| 8
| 19
|
| ANGINA
| 0
| 6
|
| CONGESTIVE HEART FAILURE
| 0
| 3
|
| ARRHYTHMIA
| 0
| 3
|
| SUPRAVENTRICULAR TACHYCARDIA
| 0
| 3
|
| MYOCARDIAL INFARCTION
| 0
| 3
|
| DEEP VENOUS THROMBOSIS
| 1
| 3
|
| PHLEBITIS
| 1
| 3
|
| TRANSIENT ISCHEMIC ATTACK
| 1
| 0
|
| ANEURYSM
| 1
| 0
|
| CEREBROVASCULAR ACCIDENT
| 0
| 3
|
| MUSCULOSKELETAL
| 7
| 16
|
| MYALGIA
| 4
| 16
|
| OSTEOPOROSIS
| 2
| 0
|
| ARTHRALGIA
| 1
| 0
|
| TUMOR LYSIS SYNDROME
| 1
| 0
|
More than 3,000 adult patients received fludarabine in studies of other leukemias, lymphomas, and other solid tumors. The spectrum of adverse effects reported in these studies was consistent with the data presented above.
Overdosage
High doses of Fludarabine Phosphate Injection, USP (see WARNINGS section) have been associated with an irreversible central nervous system toxicity characterized by delayed blindness, coma, and death. High doses are also associated with severe thrombocytopenia and neutropenia due to bone marrow suppression. There is no known specific antidote for Fludarabine Phosphate Injection, USP overdosage. Treatment consists of drug discontinuation and supportive therapy.
Fludarabine Dosage and Administration
Usual Dose
The recommended adult dose of Fludarabine Phosphate Injection, USP is 25 mg/m 2 administered intravenously over a period of approximately 30 minutes daily for five consecutive days. Each 5 day course of treatment should commence every 28 days. Dosage may be decreased or delayed based on evidence of hematologic or nonhematologic toxicity. Physicians should consider delaying or discontinuing the drug if neurotoxicity occurs.
A number of clinical settings may predispose to increased toxicity from Fludarabine Phosphate Injection, USP. These include advanced age, renal insufficiency, and bone marrow impairment. Such patients should be monitored closely for excessive toxicity and the dose modified accordingly.
The optimal duration of treatment has not been clearly established. It is recommended that three additional cycles of Fludarabine Phosphate Injection, USP be administered following the achievement of a maximal response and then the drug should be discontinued.
Renal Insufficiency
Adult patients with moderate impairment of renal function (creatinine clearance 30 to 70 mL/min/1.73 m 2) should have a 20% dose reduction of Fludarabine Phosphate Injection, USP. Fludarabine Phosphate Injection, USP should not be administered to patients with severely impaired renal function (creatinine clearance less than 30 mL/min/1.73 m 2).
Preparation of Solutions
Fludarabine Phosphate Injection, USP: Each mL contains 25 mg fludarabine phosphate, 25 mg mannitol, water for injection, q.s.; and sodium hydroxide to adjust the pH to 6.8. The pH range for the final product is 6.0 to 7.1. In clinical studies, the product has been diluted in 100 cc or 125 cc of 5% dextrose injection USP or 0.9% sodium chloride USP.
Fludarabine Phosphate Injection, USP contains no antimicrobial preservative and thus should be used within 8 hours of initial entry. Care must be taken to assure the sterility of prepared solutions. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration.
Handling and Disposal
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration.
Procedures for proper handling and disposal should be considered. Consideration should be given to handling and disposal according to guidelines issued for cytotoxic drugs. Several guidelines on this subject have been published. 1–8 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
Caution should be exercised in the handling of Fludarabine Phosphate Injection, USP. The use of latex gloves and safety glasses is recommended to avoid exposure in case of breakage of the vial or other accidental spillage. If the solution contacts the skin or mucous membranes, wash thoroughly with soap and water; rinse eyes thoroughly with plain water. Avoid exposure by inhalation or by direct contact of the skin or mucous membranes.
How is Fludarabine supplied
Fludarabine Phosphate Injection, USP is supplied as a clear, sterile solution. Each mL contains 25 mg of fludarabine phosphate, 25 mg of mannitol, water for injection, q.s.; and sodium hydroxide to adjust pH to 6.8. The pH range for the final product is 6.0 to 7.1.
| Product No.
| NDC No
.
|
|
| 109002
| 63323-192-02
| Fludarabine Phosphate Injection, USP, 50 mg per 2 mL (25 mg per mL) in a 2 mL single dose vial, packaged individually.
|
REFRIGERATE AT: 2° to 8°C (36° to 46°F).
The container closure is not made with natural rubber latex.
References
- ONS Clinical Practice Committee. Cancer Chemotherapy Guidelines and Recommendations for Practice. Pittsburgh, Pa: Oncology Nursing Society. 1999:32-41.
- Recommendations for the Safe Handling of Parenteral Antineoplastic Drugs. Washington, DC; Division of Safety, Clinical Center Pharmacy Department and Cancer Nursing Services, National Institute of Health; 1992. US Department of Health and Human Services, Public Health Service Publication NIH 92-2621.
- AMA Council on Scientific Affairs. Guidelines for Handling Parenteral Antineoplastics. JAMA. 1985;253:1590-1591.
- National Study Commission on Cytotoxic Exposure — Recommendations for Handling Cytotoxic Agents. 1987. Available from Louis P. Jeffrey, Sc.D., Chairman, National Study Commission on Cytotoxic Exposure, Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Avenue, Boston, MA 02115.
- Clinical Oncological Society of Australia: Guidelines and Recommendations for Safe Handling of Antineoplastic Agents. Med J Australia. 1983;1:426-428.
- Jones, R.B, Frank R, Mass T. Safe Handling of Chemotherapeutic Agents: A Report from the Mount Sinai Medical Center. CA Cancer Clin. 1983; 33:258-263.
- American Society of Hospital Pharmacists. ASHP Technical Assistance Bulletin on Handling Cytotoxic and Hazardous Drugs. Am J Hosp Pharm. 1990; 47:1033-1049.
- Controlling Occupational Exposure to Hazardous Drugs (OSHA Work-Practice Guidelines). Am J Health-Syst Pharm. 1996; 53:1669-1685.

PACKAGE LABEL - PRINCIPAL DISPLAY - Fludarabine 2 mL Single Dose Vial Label
Fludarabine Phosphate
Injection, USP
50 mg per 2 mL
(25 mg per mL)
For intravenous use only.
Preservative free.
Cytotoxic agent.
2 mL
Single Dose Vial Rx only

PACKAGE LABEL - PRINCIPAL DISPLAY - Fludarabine 2 mL Single Dose Vial Carton Panel
Fludarabine Phosphate Injection, USP
50 mg per 2 mL
(25 mg per mL)
For intravenous use only.
Preservative free.
Cytotoxic agent.
Refrigerate
Rx only
2 mL
Single Dose Vial

| FLUDARABINE
fludarabine phosphate injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Fresenius Kabi USA, LLC (608775388) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Fresenius Kabi USA, LLC | 023648251 | manufacture(63323-192) | |
More about fludarabine
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (1)
- Side effects
- Dosage information
- During pregnancy
- Drug class: antimetabolites
- En español
Patient resources
- Fludarabine injection drug information
- Fludarabine (Intravenous) (Advanced Reading)
- Fludarabine (Oral) (Advanced Reading)
